Klonale Eosinophilie: Erkennen und einordnen
XTRA-ARTIKEL AUSGABE 1/2020
In der Regel rufen reaktive Ursachen eine Eosinophilie hervor. Kann man diese ausschließen, muss an eine klonale Eosinophilie gedacht werden und eine zyto-/molekulargenetische Abklärung wird nötig
Text: Reinhild Herwartz
Die Eosinophilie bezeichnet eine Vermehrung der Eosinophilen, die den Absolutwert von 0,5 G/l Blut überschreitet. Während die Nachweisbarkeit mittels Blutbilddifferenzierung einfach ist, stellt die diagnostische Zuordnung eine Herausforderung dar. In 80 bis 95 Prozent der Fälle sind reaktive Ursachen wie parasitäre oder bakterielle Infekte, allergische Erkrankungen, Dermatitiden oder Autoimmunerkrankungen verantwortlich. Darüber hinaus findet man begleitende Eosinophilien bei malignen Erkrankungen der Hämatopoese wie Morbus Hodgkin, akuten Leukämien oder anderweitigen nicht hämatologischen Erkrankungen. In seltenen Fällen kann eine Eosinophilie jedoch klonal (maligne) sein. Ursächlich liegt hierbei eine klonale Stammzellerkrankung zugrunde1.
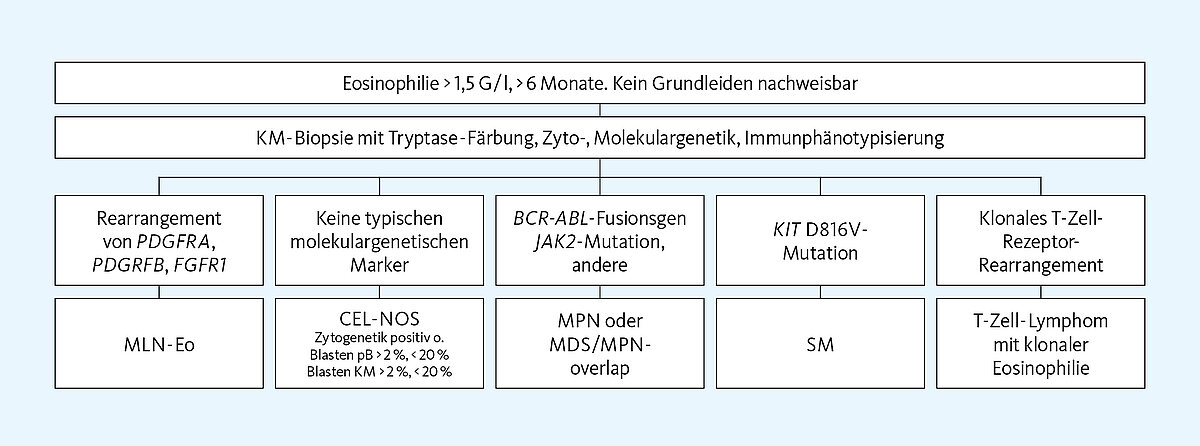
Eine klonale Eosinophilie rückt dann in den Fokus, wenn reaktive Ursachen ausgeschlossen wurden. Neben der Zytologie von Blut- und Knochenmark sowie der Histologie werden hierzu die Zyto- und Molekulargenetik erforderlich.
Das Ausmaß der Eosinophilie ist bei der klinischen und diagnostischen Bewertung ausschlaggebend und bestimmt den Schweregrad (s. Tab. 1) Bei mäßigen bis schweren Eosinophilien können, unabhängig von deren Ursache, klinische Symptome und/oder Organschäden auftreten. Hierbei sind besonders Juckreiz (Pruritus), Hautveränderungen (Urtikaria), Fieber und Schwäche zu nennen. In schweren Fällen treten Organschäden mit Organdysfunktionen auf, die mit Durchfall, Asthma, neurologischen Ausfällen oder Beteiligung des Herzens einhergehen2.

Grade der Eosinophilie
- Hypereosinophilie (HE): Persistiert die Eosinophilie über einen Zeitraum von mehr als sechs Monaten und erreicht einen Absolutwert von > 1,5 G/l, spricht man vor einer Hypereosinophilie (HE)2. Dieser Befund ist immer kontroll- und abklärungsbedürftig.
- Idiopathische Hypereosinophilie (IHE): Bleibt die Ursache der HE unklar, spricht man von einer idiopathischen Hypereosinophilie (IHE). Diese ist definitionsgemäß ohne Organbeteiligung.
- Hypereosinophiles Syndrom (HES): Liegt neben einer HE auch eine Organbeteiligung vor, die durch die Eosinophilie bedingt ist und mit Durchfall, Asthma, neurologischen Symptomen oder Beteiligung des Herzens einhergeht1, handelt es sich um ein hypereosinophiles Syndrom (HES).
Eine Eosinophilie kann selbst Ausdruck einer klonalen Stammzellerkrankung sein oder dem malignen Klon einer hämatologischen Neoplasie angehören, wenn eine frühe myeloische Stammzelle mutiert ist, die im Weiteren eine eosinophile Differenzierung durchläuft. Dann findet sich die krankheitsspezifische Genmutation auch in den Eosinophilen, woraus die Klonalität der Eosinophilie resultiert. Die Erkrankungen lassen sich wie folgt einteilen:
- Myeloische/lymphatische Neoplasie mit klonaler Eosinophilie (MLN-Eo)
- Chronische eosinophile Leukämie, nicht anderweitig zugeordnet (CEL-NOS)
- Myeloproliferative Neoplasien (MPN) oder MDS/MPN Overlap-Syndrome, bei Vorliegen bestimmter Genveränderungen, besonders: BCR-ABL, JAK2-Mutation
- Systemische Mastozytose (SM) mit KIT D816V-Mutation
- T-Zell-Lymphom mit klonaler Eosinophilie
- AML mit inv 16 oder t (16;16) (p13.1;q22)
Die vorliegende Genmutation bestimmt dabei die Beteiligung der Eosinophilen am malignen Geschehen sowie den Phänotyp der Erkrankung.
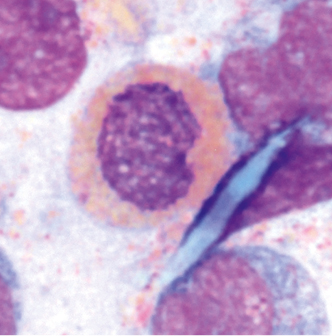
Morphologie: Bei einer klonalen Eosinophilie erscheinen die Eosinophilen oft dysplastisch verändert gegenüber reaktiven Eosinophilien. Dies ist jedoch nur indikativ und nicht als Beweis zu werten. Es können auch reaktive Eosinophilien mit morphologischen Veränderungen einhergehen sowie unauffällige Eosinophilen einer malignen Erkrankung entstammen.
Abnormitäten/Dysplasien der Eosinophilen sind:
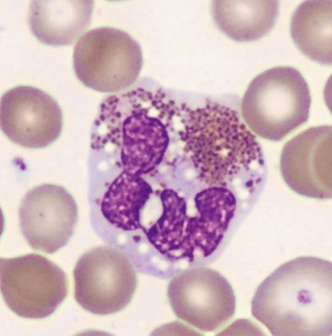
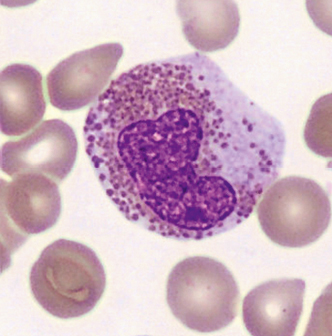
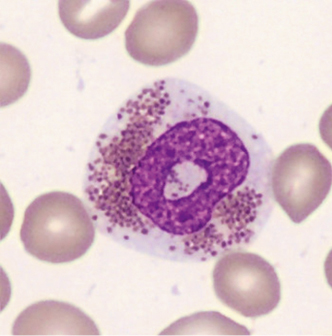
- Granulationsstörungen mit granulationsfreien Arealen, kleineren oder unreifen blauen Granula (Abb. 1–3)
- zytoplasmatische Vakuolisierungen (Abb. 1)
- Kernatypien wie Hyper- oder Hyposegmentierungen, ringförmige Kerne (Abb. 3)
- Vorkommen vergrößerter oder unreifer Eosinophilen (Abb. 2)
Im Knochenmark können sogenannte Charcot-Leyden-Kristalle als Abbauprodukte der eosinophilen Granula vorkommen. Sie sind in der Pappenheimfärbung rot-violett bis hellblau und an den Enden spitz zulaufend (Abb. 4).
1 Myeloische/lymphatische Neoplasie mit Eosinophilie
Die Charakteristika der klonalen Eosinophilie treffen im Besonderen auf die Myeloische/lymphatische Neoplasie mit klonaler Eosinophilie (MLN-Eo) zu. Bei dieser Gruppe handelt es sich um seltene maligne Erkrankungen der Hämatopoese mit klonaler Eosinophilie. Sie sind molekulargenetisch definiert und weisen entweder ein Fusionsgen oder eine Mutation auf. Je nach der vorliegenden Genveränderung unterteilt man die MLN-Eo in drei Gruppen und eine provisorische Entität [WHO-Klassifikation 2016 Arber DA et al. Blood 2016;127: 2391–2405]
- 1.1 MLN-Eo mit PDGRFA-Rearrangement (Platelet derived growth factor alpha)
- 1.2 MLN-Eo mit PDGRFB-Rearrangement (Platelet derived growth factor beta)
- 1.3 MLN-Eo mit FGFR1-Rearrangement (Fibroblasten growth factor receptor 1)
- 1.4 MLN-Eo mit PCM1-JAK2-Mutation (provisorische Entität)
Infolge der Genveränderung kommt es zur überschießenden Expression einer Tyrosinkinase mit der Folge einer Eosinophilie. Die Ausprägung der Neoplasie in Blut und Knochenmark ist heterogen. Ein Teil geht mit einer persistierenden Eosinophilie einher, man spricht dann von einer chronischen eosinophilen Leukämie (CEL). Darüber hinaus können MLN-Eo auch als lymphatische Neoplasie oder akute myeloische Leukämie auftreten. Die Erkennung der klonalen Eosinophilie ist für den Patienten therapie- und prognoseentscheidend. Es wird aufgrund der krankheitsauslösenden Mechanismen und Vorliegen der MLN-Eo eine Therapie mit einem Tyrosinkinaseinhibitor durchgeführt, auch wenn die klonale Eosinophilie als AML erscheint. Zu diesen Fällen sind die Ansprechraten gut.
1.1 MLN-Eo mit PDGRFA-Rearrangement
Bei dieser häufigsten Form der MLN-Eo liegt ein Fusionsgen FIP1L1-PDGRFA vor. Die Erkrankung tritt meist als chronische eosinophile Leukämie (CEL), selten als akute myeloische Leukämie (AML) oder T-lymphoblastische Leukämie (T-ALL) mit Eosinophilie in Erscheinung. Es erkranken auffällig mehr Männer als Frauen (17:1), meist im mittleren Lebensalter. Im peripheren Blut dominiert eine Eosinophilie, überwiegend reifzellig mit einem kleinen Anteil eosinophiler Myelozyten und Promyelozyten sowie zum Teil Abnormitäten/Dysplasien der Eosinophilen. Anämie und Thrombopenie können vorliegen. Das Knochenmark stellt sich hyperplastisch dar mit Eosinophilie, unreifen Eosinophilen und gegebenenfalls Vorkommen von Charcot-Leyden-Kristallen (Abb. 4). Die Blastenzahl kann vermehrt sein. Bei phänotypischer AML finden sich neben der Blastenvermehrung Eosinophilenwerte zwischen 1,4 und 17,2 × 103/μl1.
1.2 MLN-Eo mit PDGRFB-Rearrangement
Bei dieser sehr seltenen Form entsteht infolge des rearrangierten PDGRFB-Gens ein abnormes Fusionstranskript, meist vom Typ ETV6-PDGRFB. Im Phänotyp präsentiert sich die Erkrankung häufig als chronisch myelomonozytäre Leukämie (CMML) mit Monozytose, Dysplasiezeichen der Neutrophilen und Eosinophilie. Seltener präsentiert sich eine atypische chronisch myeloische Leukämie (aCML) ohne Philadelphia-Chromosom. Sehr selten tritt eine AML mit Eosinophilie auf.
1.3 MLN-Eo mit FGFR1-Rearrangement
Hierbei handelt es sich um eine sehr heterogene Erkrankungsgruppe. Das FGFR1-Gen auf Chromosom 8 geht hier über Translokationen Fusionen mit unterschiedlichen Partnergenen ein. Im Phänotyp können neben myeloproliferativen rscheinungsformen oder einer AML auch lymphatische Neoplasien (Lymphome oder ALL) auftreten. Insbesondere bei T-ALL oder T-lymphoblastischem Lymphom mit Eosinophilie sollte an diesen Erkrankungstyp gedacht und eine molekulargenetische Untersuchung veranlasst werden.
1.4 MLN-Eo mit PCM1-JAK2-Mutation
Zurzeit ist dies eine provisorische Entität mit der Gemeinsamkeit eines PCM1-JAK2-Fusionsgens. Im Phänotyp können MPN oder MDS/MPN-Overlap-Syndrome mit Eosinophilie und Neutrophilie vorkommen. Nicht selten besteht eine markante Dysplasie der roten Reihe. Diese Veränderung zeichnet sich durch eine hohe Rate an Transformation in eine AML aus.
2 Chronische eosinophile Leukämie
Die chronische eosinophile Leukämie (CEL-NOS) wird nach WHO 2016 den MPN zugeordnet. Es handelt sich um eine systemische Erkrankung mit persistierender autonomer Eosinophilie > 1,5 G/l im Blut, die zudem das Knochenmark und weitere Organe befällt. CEL-NOS ist immer eine Ausschlussdiagnose, die vorliegt, wenn die MLN-Eo-typischen Rearrangierungen PDGRFA, PDGRFB, FGFR1 und PCM1-JAK2 negativ sind, aber eine klonale zytogenetische Aberration gefunden wird und/oder eine Blastenvermehrung zwischen fünf und 20 Prozent in Blut oder Knochenmark vorliegt. Durch die damit bewiesene Klonalität hebt sich CEL-NOS vom HES ab, das weder eine Blastenvermehrung noch eine Genveränderung aufweist.
3 Myeloproliferative Neoplasien (MPN) oder MDS/MPN-Overlap-Syndrome mit Eosinophilie
Ein Teil der MPN oder MDS/MPN-Overlap-Syndrome geht in Abhängigkeit von der Genmutation mit klonaler Eosinophilie einher. Bei Vorhandensein des BCR-ABL-Fusionsgens ergibt sich das Bild einer CML, bei Vorliegen einer JAK2-Mutation kann sich im Phänotyp eine PMF, ET oder PV, sehr selten
CMML darstellen. Neben den erkrankungsdefinierenden Merkmalen findet sich häufig eine ausgeprägte Eosinophilie.
4 Systemische Mastozytose
Bei der Systemischen Mastozytose (SM) handelt es sich um eine Erkrankungsgruppe, die mit maligner und atypischer Mastzellinfiltration im Knochenmark und möglichem Organbefall einhergeht. Die Serumtryptase ist charakteristischerweise erhöht. Die hier typischerweise vorliegende KIT D816V-Mutation ist stark mit der systemischen Mastozytose assoziiert. Eine Eosinophilie ist häufig nachweisbar.
5 Klonale Eosinophilie assoziiert mit T-Zell-Lymphom
Eine klonale Eosinophilie kann zudem im Verein mit abnormen T-Lymphozyten auftreten. Die T-Lymphozyten zeigen einen aberranten Immunphänotyp, häufig CD3-/CD4+/CD8-. Zusätzlich weist ein großer Teil der Fälle ein klonales T-Zell-Rezeptor-Rearrangement auf.
6 AML mit inv (16) oder t (16;16) (p13.1;q22)
Diese Sonderform der AML, die dem zytologischen Typ AML M4Eo (FAB-Klassifikation) entspricht, weist eine abnorme Knochenmark-Eosinophile auf. Eine Hypereosinophilie liegt hier im Blut meist nicht vor. Im Unterschied zu den MLN-Eo steht nicht die Eosinophilie, sondern die AML im Vordergrund
und bestimmt die weitere Behandlung.
Eine Hypereosinophilie ist ein abklärungsbedürftiger Befund. Sind reaktive Ursachen ausgeschlossen, muss an eine klonale Eosinophilie gedacht und eine zyto-/molekulargenetische Abklärung durchgeführt werden. Hinter einer HE kann sich eine myeloproliferative oder Myeloische/lymphatische Neoplasie mit einer spezifischen Genmutation verbergen. Dies gilt auch, wenn die Eosinophilie mit zusätzlichen hämatologischen Auffälligkeiten wie einer Blastenvermehrung (zum Beispiel als AML mit Eosinophilie) einhergeht. Denn: Die Therapie erfolgt subtypenspezifisch. Insbesondere die MLN-Eo werden je nach zyto- und molekulargenetischem Befund mit einem Tyrosinkinaseinhibitor behandelt und zeigen darunter oft ein gutes und anhaltendes Ansprechen.
Die Autorin
Reinhild Herwartz arbeitet als Biomedizinische Fachanalytikerin Hämatologie in der Klinik für Onkologie/Hämatologie und Stammzelltransplantation der Uniklinik RWTH Aachen
Fotoquelle: Sysmex, Reinhild Herwartz
Literatur
- Swerdlow SH et al. WHO classification of Tumours of Haematopoietic and Lymphoid Tissues, Lyon 2017, S. 54–56; S. 72–79
- Fuchs R, Staib P , Brümmendorf T. Manual Hämatologie, 29. Auflage, Nora Verlag 2019, S. 347 – 353
- Arber DA et al. Blood 2016;127: 2391–2405
- www.allgemeinarzt-online.de/atemwege/a/hypereosinophilie-begleitphaenomen-oder-neoplasie-1822551
- www.onkopedia.com/de/wissensdatenbank/wissensdatenbank/eosinophilie/Eosinophilie.pdf
- Reiter A, Gotlip J. Myeloid neoplasm with Eosinophilia, Blood 2017, Volume 129, Issue 6, Page(s) 704–714
- www.onkopedia.com/de/onkopedia/guidelines/eosinophilie-assoziierte-myeloproliferative-erkrankungen-mpn-eo/@@guideline/html/index.html